No Sleep During the Reproducibility Session
Abstract
R code is provided for implementing a statistical method by Nishiura, Miyamatsu, and Mizumoto (2016) to assess when to declare the end of an outbreak of a person-to-person transmitted disease. The motivating example is the MERS-CoV outbreak in Korea, 2015. From a greater perspective, the blog entry is an attempt to advocate for spicing up statistical conferences by a reproducibility session.
 This work is licensed under a Creative Commons
Attribution-ShareAlike 4.0 International License. The
markdown+Rknitr source code of this blog is available under a GNU General Public
License (GPL v3) license from github.
This work is licensed under a Creative Commons
Attribution-ShareAlike 4.0 International License. The
markdown+Rknitr source code of this blog is available under a GNU General Public
License (GPL v3) license from github.
Introduction
A few weeks ago I went to the International Biometric Conference (IBC) in Victoria. Conferences are good for meeting people, but with respect to scientific content, there is typically no more than 2-3 talks in a week, which you really remember. Partly, this is due to the format of statistics conferences not developing much in recent decades: it is plenary talks, invited sessions, contributed sessions, showcase sessions and poster sessions all over. However, some developments have occurred, e.g.
- the German joint statistical meeting introduced the concept of a stats bazaar talk.
- the R User Conference has added some interesting additional formats, e.g. lightning talks, in order to make life at a conference more interesting. Thomas Leeper has written an inspiring blog post about this issue.
Not all science is ‘fun’, but when balancing between adding yet-another-table-from-a-simulation-study against 95% of the audience dozing off, I urge you to aim for an awake audience.
So here is an additional session format in the spirit of reproducible science, which might help make statistics conference more alive again: Take the contents of a talk, find the corresponding paper/technical report/slides, download the data (of course these are available) and start implementing. After all, hacking a statistical method is the best way to understand it and reproducing the results of an analysis is a form of peer-review we should do much more as statisticians. The important talk by Keith A. Baggerly about reproducibility in bioinformatics more than underlines this.
As a consequence, this blog entry is my attempt of a repro-session in connection with the IBC: The talk entitled Determining the end of an epidemic with human-to-human transmission by Hiroshi Nishiura was both interesting, from a field I’m interested in (infectious disease epidemiology) and the method looked like it could be re-implemented in finite time. The question the method tries to answer is the following: at which time point can one declare an outbreak of a person-to-person transmitted disease as having ended? Answering this question can be important in order to calm the population, attract tourists again, export goods or reduce alertness status. The current WHO method for answering the question requires that a period of two times the longest possible incubation time needs to have passed since the last cases before an outbreak can be declared as being over. However, as stated in their paper (Nishiura, Miyamatsu, and Mizumoto (2016)), the criterion clearly lacks a statistical motivation. As an improvement Nishiura and co-workers formulate a statistical criterion based on the serial interval distribution and the offspring distribution.
In what follows we shall quickly describe their method and apply it to their motivating example, which was the 2015 MERS-CoV outbreak in Korea. As a small outlook, we shall implement some own thoughts on how to answer the posed questions using a hierarchical model.
Method
Let \(Y_t\) be a count variable representing the number of symptom onset in cases we observe on a given day \(t\) during the outbreak. The sequence of the \(Y_t\) is also called the epidemic cuve of the outbreak. Furthermore, let \(D=\{t_i; i=1,\ldots,n\}\), be the currently available outbreak data containing the time of symptom onset in in each of the \(n\) cases of the outbreak. In what follows we will be interested in what happens with \(Y_t\) for future time points, i.e. time points after the last currently observed onset time. In particular we will be interested in, whether we will observe zero cases or more than zero cases.
The important result of Nishiura, Miyamatsu, and Mizumoto (2016) is that the probability \(\pi_t = P(Y_t > 0\>|\>D)\) can be computed as follows: \[ \begin{align*} \pi_t = 1 - \prod_{i=1}^n \sum_{o=0}^{\infty} f_{\text{offspring}}(o; R_0, k) \cdot \left[ F_{\text{serial}}(t-t_i) \right]^{o}, \end{align*} \] where \(f_{\text{offspring}}\) denotes the PMF for the number of secondary cases one primary case induces. It is assumed that this distribution is negative binomial with expectation \(R_0>0\) and clumping parameter \(k>0\). In other words, \(\operatorname{E}(O)=R_0\) and \(\operatorname{Var}(O)=R_0 + R_0^2/k\). Furthermore, \(F_{\text{serial}}\) denotes the CDF of the serial interval distribution of the disease of interest. The serial interval is the time period between the onset of symptoms in the primary and onset of symptoms in the secondary case, see Svensson (2007).
Once \(\pi_t\) is below some pre-defined threshold \(c\), say \(c=0.05\), one would declare the outbreak to be over, if no new cases have been observed by time \(t\). In other words: \[ T_{\text{end}} = \min_{t>t^*} \{ \pi_t < c \}. \] where \(t^* = \max_{i=1,\ldots,n} t_i\), i.e. the onset time in the last observed case.
Note that the formulated approach is conservative, because every available case is treated as having the potential to generate new secondary cases according to the entire offspring distribution. In practice, however, observed cases towards the end will be secondary cases of some of the earlier cases. Hence, these primary cases will be attributed as having the ability to generate more secondary cases than they actually have in practice. Another important assumption of the method is that all cases are observed: no asymptomatic cases nor under-reporting is taken into account.
Data from the MERS-Cov Oubtreak in Korea, 2015
The data basis for our analysis is the WHO data set on the MERS-Cov outbreak in Korea, which occurred during May-July 2015. It contains the information about 185 cases of the MERS-CoV outbreak in Korea, 2015. These were already analysed in a previous blog entry for the purpose of nowcasting. However, we shall now be interested in answering the following question: Given the observations of symptoms on the last (known) case on 2015-07-02. How many days without new infections would have to pass, before we would declare the outbreak as having ended?
Results
In what follows we shall distinguish results between model parameters to be estimated from data and the computation of the probability \(\pi_t\). Focus of this blog entry is on the later part. Details on the first part is available in the code.
Parameter Estimation
The parameters to estimate are the following:
- parameters of the parametric distributional family governing the serial interval distribution (in Nishiura, Miyamatsu, and Mizumoto (2016) this is assumed to be a gamma distribution)
- parameters of the offspring distribution, which here is assumed to be negative binomial with mean \(R_0\) and clumping parameter \(k\)
The first step is easily accomplished in Nishiura et al. (2015) by solving for given mean and standard deviation for the serial interval distribution observed in secondary data - see the paper for details. The solution can be found analytically given the values.
E <- 12.6
SD <- 2.8
(theta_serial <- c(E^2/SD^2,E/SD^2))## [1] 20.25 1.61The second part is addressed in Nishiura et al. (2015) by
analysing final-size and generation data using a maximum likelihood
approach. We will here only implement the methods using the data
presented in Figure 1 and Table 1 of the paper. Unfortunately, one
cluster size is not immediately reconstructable from the data in the
paper, but guesstimating from the table on p.4 of the ECDC
Rapid Risk Assessment it appears to be the outbreak in Jordan with a
size of 19. The likelihood is then maximized for \(\mathbf{\theta}=(\log(R_0),\log(k))'\)
using optim. Based on the Hessian, a numeric approximation
of the variance-covariance matrix of \(\hat{\mathbf{\theta}}\) can be
obtained.
Altogether, we maximize the combined likelihood consisting of 36 as well as the corresponding number of generations by:
theta_mle <- optim(c(log(1),log(1)),ll_combine, outbreaks=outbreaks, control=list(fnscale=-1),hessian=TRUE)
exp(theta_mle$par)## [1] 0.826 0.128These numbers deviate slightly from the values of \(\hat{R}_0=0.75\) and \(\hat{k}=0.14\) reported by Nishiura et al. (2015). One explanation might be the unclear cluster size of the Jordan outbreak, here it would have been helpful to have had all data directly available in electronic form.
Outbreak End
The above \(\pi_t\) equation is
implemented below as function p_oneormore. It requires the
use of the PMF of the offspring distribution (doffspring),
which here is the negative binomial offspring distribution.
##Offspring distribution, this is just the negative binomial PMF.
doffspring <- function(y, R_0, k, log=FALSE) {
dnbinom(y, mu=R_0, size=k, log=log)
}
##Probability for one or more cases at time t.
p_oneormore <- Vectorize(function(t,R_0,k,theta_serial,yMax=1e4,verbose=FALSE) {
if (verbose) cat(paste0(t,"\n"))
res <- 1
##Loop over all cases as in eqn (1) of the suppl. of Nishiura (2016).
##Setup process bar for this action.
if (verbose) {
pb <- startpb(1, nrow(linelist))
on.exit(closepb(pb))
}
for (i in seq_len(nrow(linelist))) {
if (verbose) { setpb(pb, i) }
serial_time <- as.numeric(t - linelist$Date.of.symptoms.onset[i])
cdf <- pgamma(serial_time, theta_serial[1], theta_serial[2])
y <- 0L:yMax
ysum <- sum( doffspring(y=y,R_0=R_0,k=k)*cdf^y)
res <- res * ysum
}
return(1-res)
},vectorize.args=c("t","R_0","k"))The function allows us to re-calculate the results of Nishiura, Miyamatsu, and Mizumoto (2016):
##Results from the Nishiura et al. (2015) paper
##R_0_hat <- 0.75 ; k_hat <- 0.14
##Use MLE found with the data we were able to extract.
R_0_hat <- exp(theta_mle$par[1])
k_hat <- exp(theta_mle$par[2])
## Compute prob for one or more cases on a grid of dates
df <- data_frame(t=seq(as.Date("2015-07-15"),as.Date("2015-08-05"),by="1 day"))
df <- df %>% mutate(pi = p_oneormore(t,R_0=R_0_hat, k=k_hat, theta_serial=theta_serial, yMax=250,verbose=FALSE))
head(df, n=3)## Source: local data frame [3 x 2]
##
## t pi
## (date) (dbl)
## 1 2015-07-15 0.366
## 2 2015-07-16 0.297
## 3 2015-07-17 0.226We can embed estimation uncertainty originating from the estimation of \(R_0\) and \(k\) by adding an additional bootstrap step with values of \((\log R_0, \log k)'\) sampled from the asymptotic normal distribution. This distribution has expectation equal to the MLE and variance-covariance matrix equal to the observed Fisher information. Pointwise percentile-based 95% confidence intervals are then easily computed. The figure below shows this 95% CI (shaded area) together with the \(\pi_t\) curve.
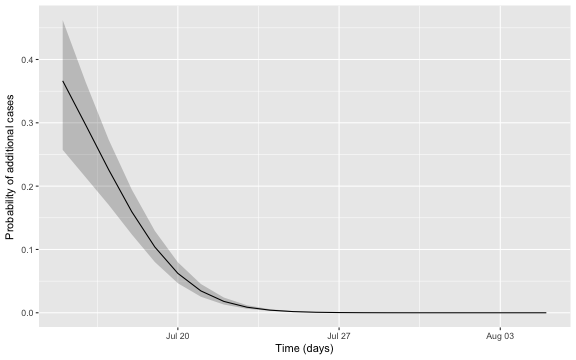
Altogether, the date where we would declare the outbreak to be over is found as:
c_threshold <- 0.05
(tEnd <- df2 %>% filter(`quantile.97.5%` < c_threshold) %>% slice(1L))## # A tibble: 1 x 4
## t pi `quantile.2.5%` `quantile.97.5%`
## <date> <dbl> <dbl> <dbl>
## 1 2015-07-21 0.0345 0.0253 0.0454In other words, given the assumptions of the model and the chosen threshold, we would declare the outbreak to be over, if no new cases are observed by 2015-07-21. The adequate choice of \(c\) as cut-off in the procedure in general depends on what is at stake. Hence, choosing \(c=0.05\) without additional thought is more than arbitrary, but a more careful discussion is beyond the scope of this blog note.
Hierarchical model
Commenting on the derivations done in Nishiura, Miyamatsu, and Mizumoto (2016) from a Bayesian viewpoint, it appears more natural to formulate the model directly in hierarchical terms:
\[ \begin{align*} N_i &\sim \operatorname{NegBin}(R_0,k), & i&=1,\ldots,n,\\ \mathbf{O}_i\>|\>N_i &\sim \operatorname{M}(N_i,\mathbf{p}_{\text{serial}}),& i&=1,\ldots,n,\\ Y_t\>|\> \mathbf{O} &= \sum_{i=1}^n O_{i,t_i-t}, & t&=t^*+1,t^*+2,\ldots,\\ \end{align*} \] where \(\mathbf{p}_{\text{serial}}\) is the PMF of the discretized serial interval distribution for exampling obtained by computing \(p_{y} = F_{\text{serial}}(y) - F_{\text{serial}}(y-1)\) for \(0<y\leq S\), where \(S\) is the largest possible/relevant serial interval to consider, and letting \(p_{0} = 0\). Furthermore, \(O_{i,t_i-t}=0\) if \(t_i-t<0\) or \(t_i-t>S\) and corresponds to the value obtained from \(M(N_i,\mathbf{p}_{\text{serial}})\) otherwise. Finally, \(\mathbf{O}=(\mathbf{O}_1,\ldots,\mathbf{O}_n)\).
Given \(R_0\) and \(k\) it is easy to use Monte Carlo
simulation to obtain instances of \(Y_t\) for a selected time-range from the
above model. The code for this function simulation is
available as part of this R-markdown document (again, see the underlying
source on the github repository for details). Similarly to the previous
model the hierarchical model is also slightly conservative, because it
does not take existing secondary cases in the data into account and
samples \(N_i\) new secondary cases for
each observed case.
Since we for this model will be using simulations it is easy to modify the criterion for fade-out slightly to the more natural probability \(\pi_t^*\) that no case at \(t\) nor beyond \(t\) will occur, i.e. \[ \pi_t^* = P\left( \bigwedge_{i=t}^\infty \{Y_t = 0\} \right). \]
We perform a study with 10000 different simulations each evaluated on
a grid from 2015-07-03 to 2015-07-27. The resulting values are stored in
the \(25 \times 10000\) matrix
Y from which we can compute:
pi <- apply(Y,1,mean)
pi[pi < c_threshold]## 2015-07-21 2015-07-22 2015-07-23 2015-07-24 2015-07-25 2015-07-26 2015-07-27
## 0.0341 0.0197 0.0095 0.0037 0.0021 0.0013 0.0004##Better way to calc extinction prob.
pi_star <- rev(apply(apply(Y,2,function(x) cumsum(rev(x))>0),1,mean))
pi_star[pi_star < c_threshold]## 2015-07-22 2015-07-23 2015-07-24 2015-07-25 2015-07-26 2015-07-27
## 0.0343 0.0168 0.0075 0.0038 0.0017 0.0004We note that the result, when using \(\pi_t^*\) instead of \(\pi_t\), leads to the outbreak being declared over one day later. Additional uncertainty handling is performed as before by obtaining bootstrap samples for \((\log R_0, \log k)'\) from the asymptotic normal distribution. For each such sample the above Monte Carlo procedure is executed allowing us to determine point-wise confidence intervals for the probability by the percentile method.

Discussion
The present note introduced the statistical model based approach of Nishiura, Miyamatsu, and Mizumoto (2016) for declaring the end of a person-to-person transmitted disease outbreak such as MERS-Cov, Ebola, etc. If the considered outbreak has a different mode of transmission, e.g. foodborne or originates from a point-source, then different formulas apply, see e.g. Brookmeyer and You (2006). Interestingly enough, there appears to be some methodological overlap between declaring the end of an outbreak and declaring a software product to be free of errors.
To summarise: The results of the Nishiura, Miyamatsu, and Mizumoto (2016) paper could - with some fiddling to guesstimate the data - be approximately reproduced. A hierarchical model with simulation based inference was able to produce similar results. Availability of the full data in electronic form would have been helpful. Altoghether, it was fun to implement the method and hope is that the avaibility of the present analysis and R code might be helpful to someone at some point. You are certainly invited to reprofy the present analysis.

Acknowledgements
I thank Hiroshi Nishiura for answering questions about their paper.